제약회사가 환자를 대상으로 3상 임상시험을 하는 조건으로 의약품 당국으로부터 급하게 개발 의약품의 시판허가를 받아놓고는 시장에서 외면받자 생산 자체를 하지 않는 경우가 다수 있는 것으로 나타났다.
4일 식품의약품안전처가 국회 보건복지위원회 장정숙 의원에게 제출한 ‘최근 3년간(2015∼2017년) 3상 임상 조건부 허가 의약품 및 생산현황’ 자료를 보면, 이 기간 총 23개 의약품이 3상 조건부 시판허가를 받았다.
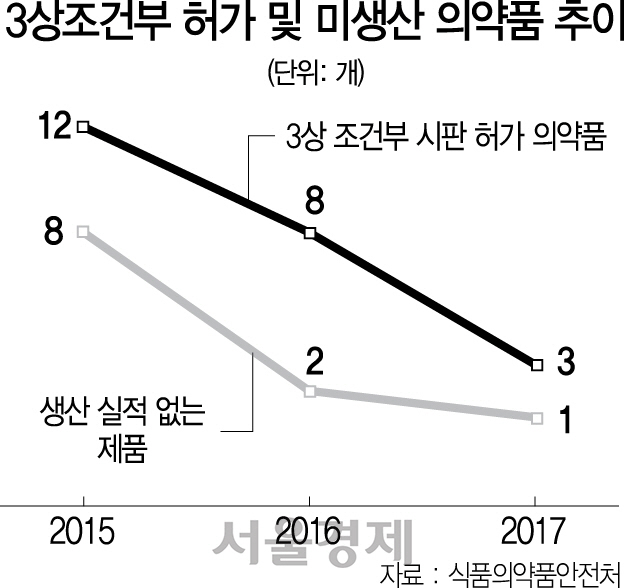
3상 조건부 허가제도는 생명을 위협하는 질병이거나 현존 치료법으로는 치료가 불가능한 경우(항암제, 희귀의약품 등) 환자에게 신속한 치료기회를 제공할 목적으로 시판 후 3상 확증 임상시험 자료 제출을 조건으로 식약처가 시판허가를 해주는 제도다. 연도별로는 2015년 12개 품목, 2016년 8개 품목, 2017년 3개 품목이 3상 조건부 시판허가를 받았다.
하지만 이렇게 위급을 다투는 환자를 위해 급하게 허가받은 3상 조건부 허가 의약품 23개 중에서 절반에 가까운 11개(47.8%)는 생산실적이 전혀 없는 것으로 드러났다. 구체적으로 코텔릭정 20㎎, 트랜스라나과립(125㎎, 250㎎, 1000㎎), 로스미르, 리아백스주, 입랜스캡슐(75㎎, 125㎎), 자이카디아캡슐(150㎎) 등이었다. 특히 자이델릭정(100㎎, 150㎎)은 생산실적이 전무한 데다 아예 자진 취소해버린 것으로 나타났다.
또 장의원측에 따르면 이 제도가 국산신약 개발을 독려하기 위해 마련한 제도임에도 23개 품목 중 국산 신약은 단 3개뿐(13%)이었다. 장 의원이 제시한 ‘의약품의 품목허가·신고·심사 규정’의 제58조 신설 당시(2008년 8월14일), 국내 개발 신약, 개량신약은 우선하여 심사토록 한다는 내용이 포함됐다.
장정숙 의원은 “환자 치료 때문에 신속 허가로 일종의 특혜를 주는 것인데, 제약회사가 허가받고도 치료제를 공급하지 않는다면 ‘조건부 허가제’의 의미는 퇴색된다”고 지적했다. 장 의원은 또 “식약처는 허가 전 수요조사, 시판 후 공급계획, 사후 조건충족 여부 등을 사전에 철저하게 조사하고, 생산하지 않는 제품은 과감히 정비해야 한다”고 그는 강조했다.









